

TLDR
Pharma won’t unlock the promised $100B from AI by bolting generic LLMs onto legacy workflows; they can’t meet pharma’s ‘X‑Factor’ of auditability, domain depth, and GxP‑grade control.
The real value comes from healthcare‑native systems of action that embed domain‑specific LLMs inside a compliance‑first infrastructure with full data lineage and deterministic workflows.
For buyers, the key question shifts from “How smart is this model?” to “Can this system survive a 21 CFR Part 11 / GxP audit and still deliver 2–5x throughput?”
The article offers a practical X‑Factor evaluation checklist so procurement, QA, and IT can separate shallow AI wrappers from pharma‑grade platforms that are truly deployable at scale.
Here’s the uncomfortable truth: generic LLMs aren’t going to unlock the $100B in annual value that pharma keeps hearing about. Not on their own, anyway. They need to live inside a unified, compliant capability layer. One that spans everything from discovery to commercialisation.
Skip that, and skip native support for 21 CFR Part 11 and audit-grade data provenance, and what you’ve got is a clever toy. Regulators and QA won’t let it anywhere near core workflows, and they’d be right not to.
The $100B opportunity. And why pilots stall
Analysts keep pointing to the same story: generative AI could reshape R&D, clinical, manufacturing, and commercial functions across life sciences, with value pools worth tens of billions a year. Deloitte and McKinsey both make the same point in different words [4] [5]. The real gains come from automating literature review, trial design, safety signal detection, and commercial analytics, but only when AI is scaled as an enterprise capability. Not a scatter of disconnected tools.
And yet, most pharma organisations are still stuck in pilot mode. Sound familiar?
- Discovery teams running point AI tools for target identification
- Clinical groups testing protocol drafting copilots
- Commercial teams trialing AI assistants for MSLs and reps
What you end up with is a patchwork of unconnected ‘AI widgets,’ all sitting on top of siloed data and legacy governance. That fragmentation is exactly what’s blocking the lifecycle-wide value Bessemer and others say is on the table. Value that only shows up when AI works as a shared, horizontal layer.
Why generic LLMs fail the pharma “X-Factor” test
Most off-the-shelf LLMs, and the generic AI wrappers built on top of them, quietly assume that “good enough text” equals value. In pharma, that assumption breaks down for three structural reasons.
Non-determinism and black-box behaviour. Ask an LLM the same question twice, and you might get two different answers. Worse, the model won’t tell you how it got there. That’s classic black-box risk [6], and in a regulated environment, it’s a serious problem. How do you prove why a recommendation, classification, or summary was produced if even the system can’t explain itself?
Lack of verifiable provenance. GenAI systems often can’t tell you which documents, data points, or versions actually shaped a given output. No lineage means no way to reconstruct the chain of evidence an auditor or safety board will demand the moment something goes wrong.
No built-in GxP controls. Generic AI tools simply weren’t built for GCP/GMP/GLP environments. Environments that require controlled vocabularies, locked records, role-based access, and validated systems as a baseline. Wrapping a consumer-grade LLM in SOPs and manual review doesn’t fix this. It just papers over it, and it won’t satisfy the letter or the spirit of pharma regulation.
A recent industry analysis on black-box models and hallucination risk in pharma put it bluntly: model opacity directly undermines audit trail completeness, data integrity, and accountability. As the authors note, this opacity creates real challenges for trust, regulatory compliance, and accountability across the industry [6].
That’s the X-Factor, really. If a system can’t explain itself in a way that holds up under FDA and EMA scrutiny, it never gets deployed where the value actually lives [2].
21 CFR Part 11: the non-negotiable foundation
Title 21 CFR Part 11 is the rulebook for when electronic records and signatures count as trustworthy. Equivalent to paper, in the FDA’s eyes [3]. It calls for validated systems, secure and time-stamped audit trails, and controls that stop records from being altered without keeping the historical version intact.
In practice, a compliant system needs to:
- Link electronic signatures to specific records, so they can’t be lifted out or reused to falsify data
- Maintain audit trails capturing who did what, when, and why. Without burying earlier versions
- Produce complete, accurate copies of records that hold up to regulatory review
Even where enforcement discretion applies in certain areas, guidance is consistent on one thing: organisations still have to meet predicate rule requirements around dates, times, sequence of events, and record integrity [1].
Here’s where generic LLMs come up short out of the box:
- They don’t natively keep immutable, time-stamped logs of prompts, context, and outputs tied to specific regulated records
- They don’t link outputs to controlled versions of source data, model versions, or human reviewers
- They don’t expose governance hooks that connect generated content to electronic signatures or approval workflows
One FDA-aligned summary puts the goal of Part 11 simply: electronic records and signatures need to be trustworthy, reliable, and generally equivalent to paper records and handwritten signatures [1]. If your AI can’t demonstrate that, it stays in the sandbox. And the $100B opportunity stays theoretical.
From black box to glass box: audit-grade provenance
Getting past pilot mode means holding AI in pharma to the same auditability bar as any other validated, GxP-relevant system. That means turning the LLM from a black box into something closer to a glass box. Where every insight has a traceable lineage.
Research on AI auditability in clinical settings points to three capabilities worth demanding from any enterprise-grade AI platform [9]:
- Data provenance: logging and exposing where every input came from, how it was transformed, and which model and configuration produced the output.
- Explainability aligned with regulatory guidance [8]: deep models are still complex, but you need a way to show how inputs relate to outputs, mapped to what regulators actually expect to see.
- Evidence of human oversight: logs that clearly show review, approval, and intervention by qualified people. Not just “the AI said so.”
A provenance-driven audit framework has already shown it can support fairness auditing and regulatory transparency in clinical AI [7]. The same logic carries over to pharma: without rich provenance logs, there’s no convincing way to show your AI-assisted workflows are actually under control.
One analysis of modern AI audits puts a name to the danger here. A “provenance gap,” where production models and data quietly drift from what was originally validated [10]. Relying on static PDF reports and one-off attestations is how that gap forms. And that gap is exactly where regulatory, safety, and reputational risk piles up.
Why the “unified capability layer” matters
The biggest strategic shift isn’t a new tool. It’s a new mental model. Stop treating AI as a stack of departmental point solutions. Start treating it as a horizontal capability layer that runs across the entire pharma value chain. Advisory firms agree on this point: real value only shows up once AI is integrated across R&D, clinical, manufacturing, and commercial as one coordinated fabric [4] [5].
In practice, a unified AI capability layer should:
- Connect discovery, preclinical, clinical, regulatory, and post-market datasets, with consistent semantics and access controls
- Provide shared services for identity, access, audit trails, and data lineage across every AI application
- Enforce consistent validation, change control, and monitoring across models, prompts, and workflows
Once that layer exists, something interesting happens. Insights from early discovery can safely flow into protocol design and site selection, which then feed safety surveillance and payer value dossiers. All without breaking the audit trail. Without it, every single use case has to reinvent governance from scratch. That’s what slows delivery down and quietly stacks up risk.
A quick way to picture it
Imagine a diagram for your next internal deck or board update:
- A central “Unified AI capability layer” node sits at the top
- Branching down: Target ID, Preclinical, Clinical Trial Design, Regulatory Submissions, Post-Market Safety
- A side node, “Compliance & data infrastructure,” feeds directly into Clinical and Regulatory
Figure 1:

The message in one line: the AI brain can be shared. It’s the compliance spine that makes it actually deployable.
What a “pharma-grade” AI platform needs to look like
For anyone making the call, the real question is simple: what has to be true about our AI platform before we scale it across the enterprise? Based on where regulatory expectations and best practice currently stand, five design principles stand out.
1. Compliance-first architecture. 21 CFR Part 11 [3], data integrity, and GxP need to be foundational design constraints. Not bolted on after the fact. Built-in system validation, access control, audit trails, and record retention, configured to match your SOPs and quality systems.
2. Native data provenance and lineage. Every AI-enabled interaction gets logged with full context: source datasets, document versions, model IDs, prompts, parameters, and downstream actions. That provenance needs to be queryable, so QA, safety, and regulators can reconstruct the decision path behind any critical output.
3. Model and prompt lifecycle management. You need formal processes, and the tooling to back them up. For versioning, validating, approving, and retiring models, prompts, and composite agents. Every change auditable, with clear links to testing, risk assessments, and sign-offs. Same as any other validated system.
4. Embedded human-in-the-loop controls. The platform needs to support risk-based human review for high-impact decisions, with approvals and rationales captured in a structured way. This isn’t just a UI nicety, it has to be built into the record and signature model itself to stay Part 11 compliant [1].
5. Enterprise integration and interoperability. To act as a true horizontal layer, the platform needs clean integration with EDC, CTMS, safety systems, document management, and commercial CRM through standardized interfaces. That’s how you avoid building “AI islands” and instead create shared context that compounds in value over time.
A simple table and chart for board-level conversations
When you’re briefing leadership, this 4×4 cuts through the hype fast:
Table 1: AI Operating Models in Pharma: Compliance Readiness vs. Lifecycle Value Capture
| AI approach type | Compliance readiness | Value capture scope | Strategic risk level |
|---|---|---|---|
| Generic LLM chatbots | Low — minimal audit trails, no Part 11 alignment | Narrow — non-GxP tasks, experimentation | High — reputational and regulatory if misused |
| Departmental point solutions | Medium — partial controls per tool | Local — R&D only or commercial only | Medium — governance fragmentation |
| Unified AI capability layer | High — native compliance and provenance | Enterprise — lifecycle-wide value capture | Managed — controlled scaling with oversight |
| Future state with on-chain provenance | Very high — cryptographically verifiable lineage | Extended — partners, ecosystem, automation | Emerging — new standards, but strong trust posture |
Figure 2: Why Unified AI Outperforms Generic LLMs in Pharma 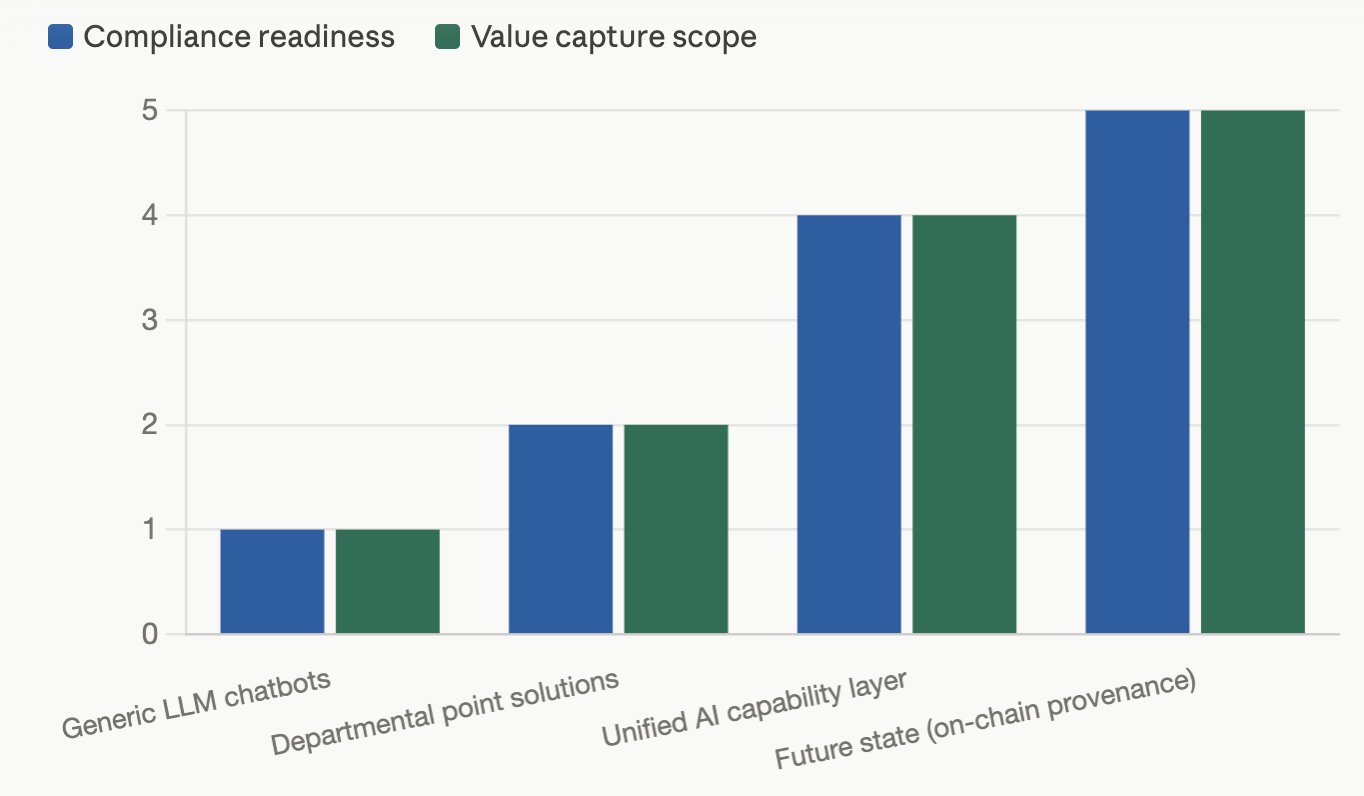
Only a unified capability layer with embedded compliance hits both high-value potential and an acceptable risk profile.
What leaders should do now
The call to action here isn’t ‘run more pilots.’ It’s ‘design the capability layer that’s going to carry us for the next decade.’
Three steps to start with:
- Define your AI governance target state. Get quality, regulatory, pharmacovigilance, and IT aligned on what ‘pharma-grade AI’ actually means for your organisation. Anchored explicitly in Part 11, data integrity, and provenance requirements.
- Select platforms on compliance and provenance, not just model quality. When you’re evaluating vendors, weight questions about audit trails, lineage, and validation artefacts just as heavily as benchmarks or latency numbers.
- Prioritise cross-lifecycle use cases. Aim your first scaled deployments at workflows that naturally bridge domains, connecting medical writing, regulatory submissions, and safety signal review, for example, to prove out the ROI of a unified layer early.
As DDReg’s “AI Auditability [10]:

Author: Stephen
Founder of HealthyData.Science · 20+ years in life sciences compliance & software validation · MSc in Data Science & Artificial Intelligence.